Submitted by ve10 on
My protein of the week, 78-residue 2J8B (CD59 glycoprotein), was folding swimmingly. Below is a typical time-series of the discrepancy:
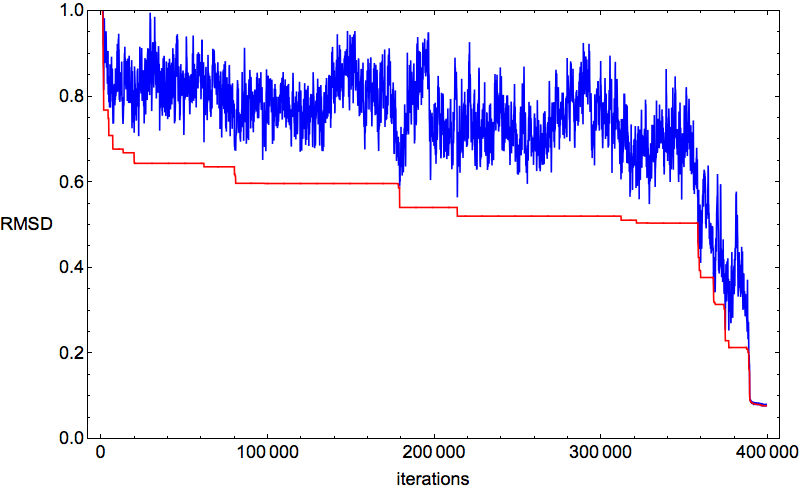
After a few hundred thousand iterations of thrashing about, click and all the constraints fall into place. This behavior of the algorithm is the best possible for the hypothesis that the constraints specify a unique fold. And in fact that was confirmed by the percentage-below-distance-cutoff plots:
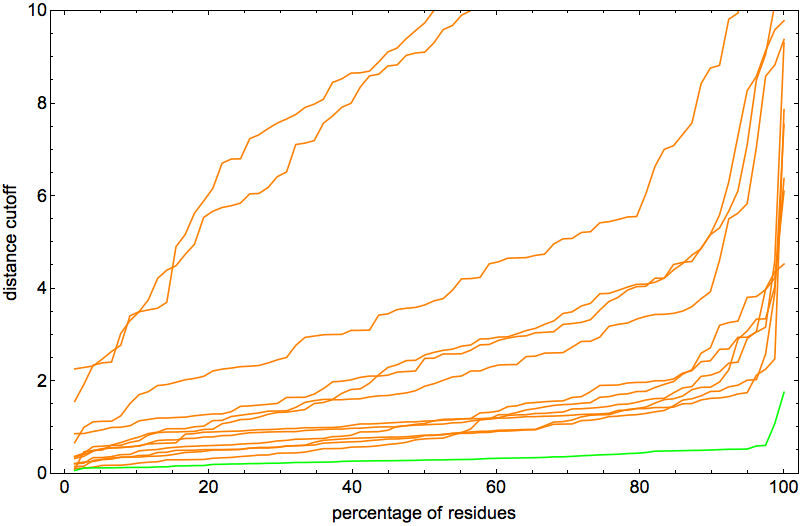
Note that the vertical scale is a factor of two smaller than in the previous posts. The two outlier curves were actually good folds too, differing from native only by rotation about a single “hinge”. In fact, I was so happy with the behavior of the algorithm on 2J8B it seemed like the perfect protein for narrowing down the optimal ADMM “discrepancy accumulation parameter”, α. The plot above corresponds to α = 0.002. With α = 0.01 I got pretty much the same (excellent) results:
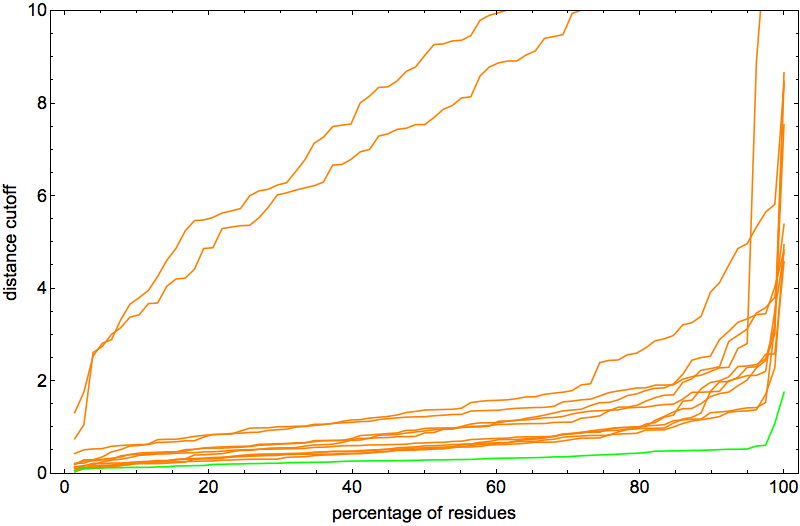
But with α = 0.05 the algorithm didn’t succeed at finding a fold at all! So now we know that α has to be small, but probably not quite as small as reported in a previous post. The median number of iterations for these two values of α were about the same, a half-million.
Over the course of the week an uneasy feeling started to take hold that maybe 2J8B was folding too well. This suspicion was dramatically made plain when Hyung Joo added a new datum to each litemotif: the PDB name of the protein from which it was extracted. All my 2J8B folding experiments were carefully avoiding native litemotifs, and Hyung Joo’s input file looked good because the identifier 2J8B, and plausible homolog names (2J8C, 3J8B, …), were absent.
Imagine my surprise, then, when I looked at the new information in the output file of the folding algorithm that reports on the sources (PDB names) of the litemotifs it was using as constraints and found a surprising abundance of the names 1CDQ, 2OFS, 2UX2, and 4BIK!
Curse you, Naming Convention Committee of the PDB! The PDB structures 1CDQ, 2OFS and 2UX2 are really the same structure, just different “deposits” of experimental results, and 4BIK contains this same structure as part of a larger structure. The small residual discrepancy in my experiments were therefore not confirming the transferability of litemotifs, but something more trivial: the consistency of various experimental structure determinations of the same stupid protein.
It’s easy to safeguard against protein name-aliases and we are going to implement that right away to avoid future embarrassments. And even with those native-contaminated input files I’m still very impressed by the power of the algorithm. Consider this: the average number of litemotifs per constraint for 2J8B was 140, or less than 3% were native (taken from the aliases 1CDQ, 2OFS, 2UX2, and 4BIK). But the algorithm doesn’t know which 3% of the choices at each of the 75 constraints are the correct ones!
We are still very good at solving protein-puzzles, but after this week, a little bit less sure about the scheme for creating these puzzles.
