Submitted by ve10 on
My routine is different this week, because of Cornell's "Spring" break. While my Analytical Mechanics students are developing a more nuanced nose for tequila at lower latitudes, I'm doing research at home, on the outer rim of Ithaca. I live 6.8 miles from my office in Clark Hall, the perfect distance for interrupting my day with a slow half-marathon office-tag in advance of the Skunk Cabbage Classic this Sunday. As a solitary runner my chief enemy is boredom. On that front I've been helped by Lionel Levine's hat puzzle, Alex Alemi's switch puzzle, and of course that bottomless chasm called protein folding.
Monday
On this, my first day of break, I decided to grab a larger chunk of the PDB to work with. Again, using the PDB advanced search interface, I broadened the X-ray resolution from 1.5 Å to 1.7 Å, and lifted the upper bound on the chain length (lower bound is still 50). Besides the original restrictions (pure protein, single chain, no ligands) I added a few more that probably made little difference: number of "entities" and "models" both set to 1, no synthetic residues. The result was 999 proteins.
Using Mathematica I quickly realized some of the PDB files were problematic. Mathematica always parses the sequence and structure of the PDB file as a list of chains, and it is true that with my setting of 1 for the number of chains on the PDB interface, 63% of my files had just one item in the list of chains. I've put off for now trying to extract local structure from the remaining 37%. When I get around to that, here are two hypotheses to explore. First, I noticed that in some of these problematic cases the PDB description actually gave multiple variants of the same basic chain. Second, when the PDB description was indeed a single chain of a given length, the combined lengths of the multiple chains parsed by Mathematica often came close to the PDB length. Perhaps this is a case of breaks being introduced when, say, the structure is poorly resolved in particular places.
Another shortcoming in some of the PDB structures is residues lacking their full complement of side-chain atoms. I suppose that the X-ray structure simply leaves out atoms that are poorly resolved. In summary, I am now working with 627 protein molecules where a small fraction of the side chain atoms, even at 1.7 Å, are absent. Since I only look at the side chain atoms to establish "contacts" between residues, and the highly mobile/disordered atoms are less likely to form contacts, I am not too worried.
Tuesday
I started the day by writing slightly more efficient Mathematica code for extracting the three local structure types I now denote by *XY*:Z, *X*Y*:Z and *X**Y*:Z. The letters X, Y, Z represent residues whose side-chain atoms form mutual contacts, where X and Y are local on the backbone, separated by 0, 1 or 2 "wildcards", while Z is anywhere on the chain. The structures associated with these types are defined by 5, 6 and respectively 7 α-carbon atom positions. These are the only possibilities when there are just three "named" residues, two of which are backbone-local, and the local backbone structure extends at most 1 residue away from the named residues.
Here are the tallies for the numbers of local structures extracted from my 627 proteins:
*XY* : Z 14711
*X*Y* : Z 21563
*X**Y* : Z 14518
The most popular in these categories are *YC*:T, with 51 instances, *V*L*:L, with 115, and the runaway champion, *L**L*:L with 267.
Looking ahead to the application of these structures as constraints in the folding algorithm, I was curious what fraction of residues, in any particular protein, are participants in one of my local structures. Here is the histogram of the "participation fraction":
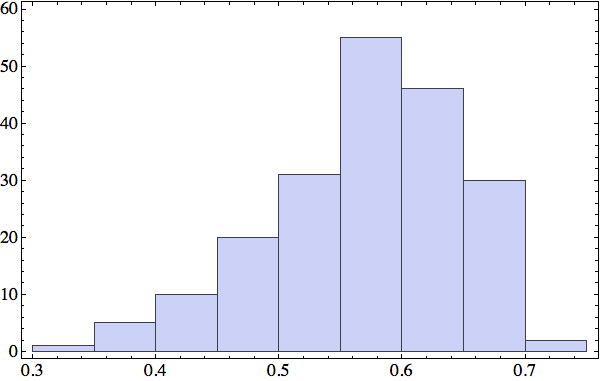
I will have to look closely at the proteins at the low end of this distribution. For those at the high end there probably are enough constraints to determine a unique fold. In any case, I've got a number of options for increasing the number of constraints. One is my cutoff on the distance between side-chain atoms that defines residue contacts. All the data in this post is based on a 5 Å cutoff. My side-chain distance histogram from last week showed a second peak at 5.5 Å that might correspond to another type of contact that I am currently ignoring.
Wednesday
Today I'm too sore to run a half-marathon; I will instead make pictures of local structures, the constraints for my folding algorithm. I've set this up in Mathematica so I can retrieve all the local structures for say, *YC*:T, and render the 4+1 α-carbons of all of them as a short animation. Using rbProj I translate and rotate all of these so they are as close as possible to one of them. For most of the structure codes I've looked at, and *YC*:T (animated below) is a good example, these animations are consistent with a unique structure:
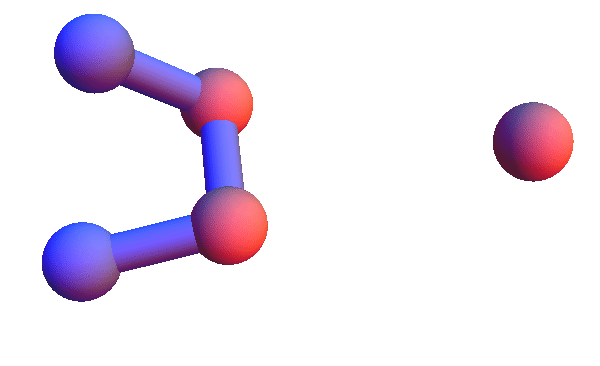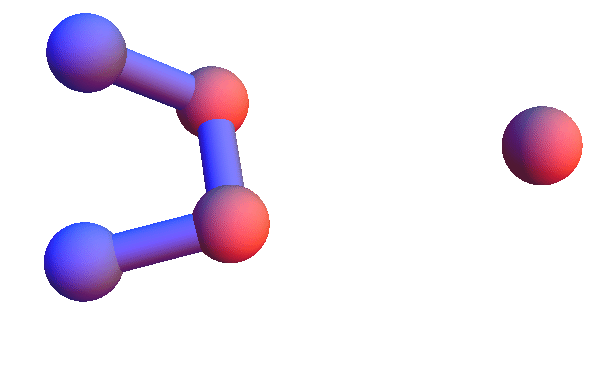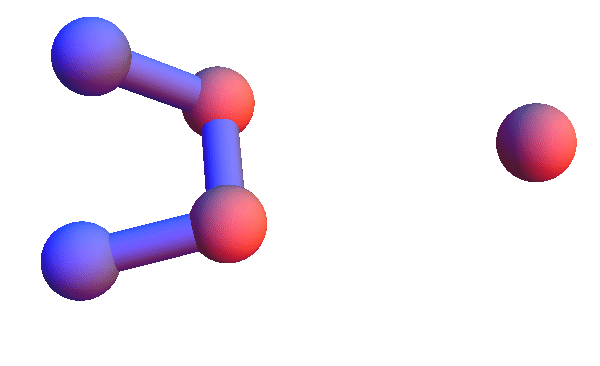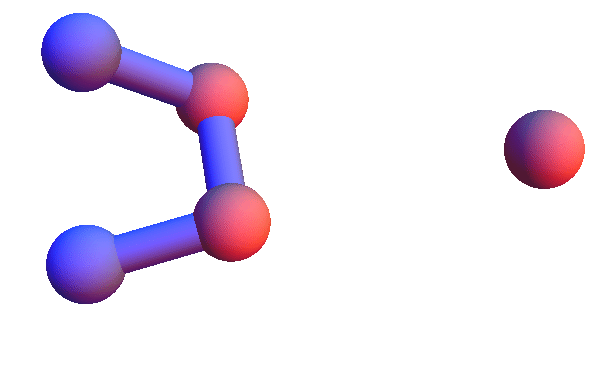
I've colored the trio of residues making mutual contacts in red, since that property is not at all obvious when the side chains are not shown. Each frame in my animations is at least within 0.4 Å root-mean-square deviation (RMSD) in its atom positions from some other frame. All the 4+1 structures I've looked at have alpha-helix character. When we go to the 5+1 class, the backbones tend to be un-twisted, as in beta-sheets. Here is an animation for *H*L*:I :
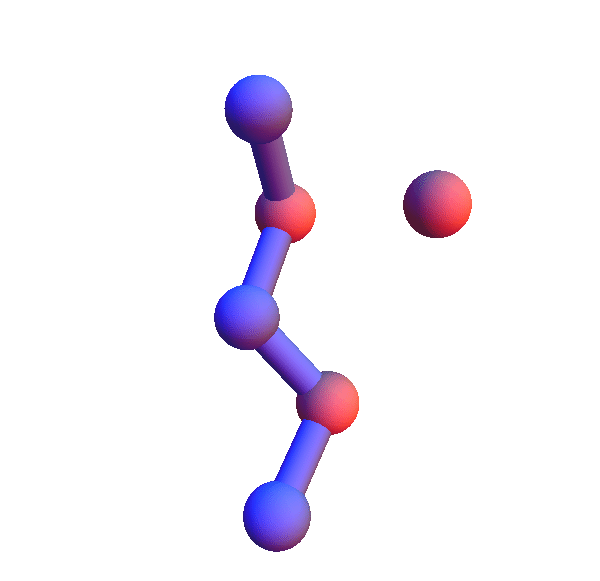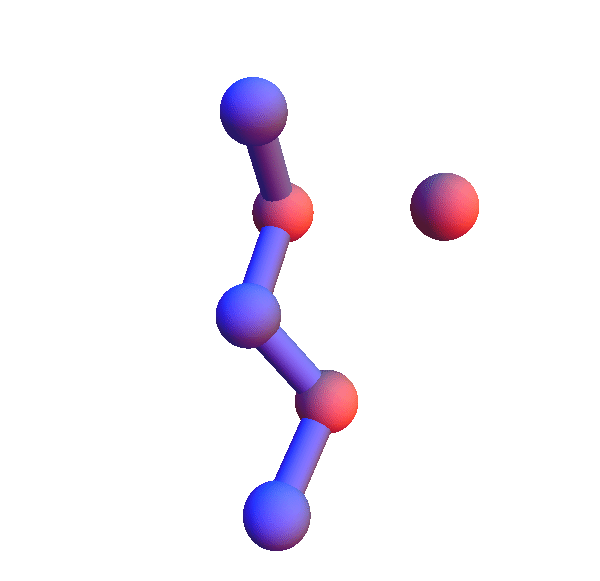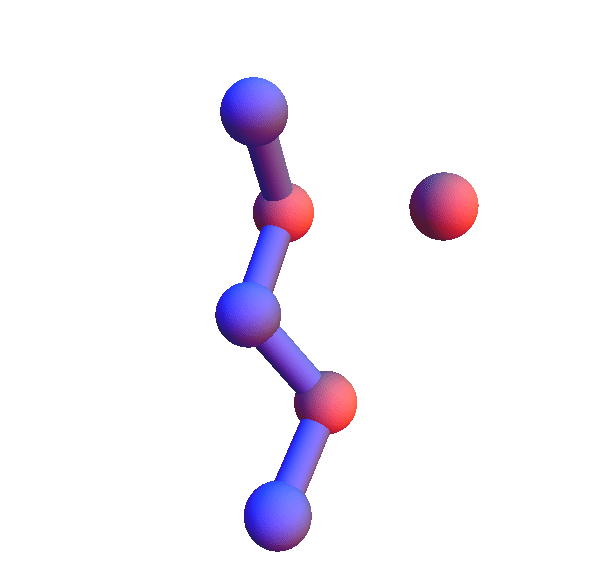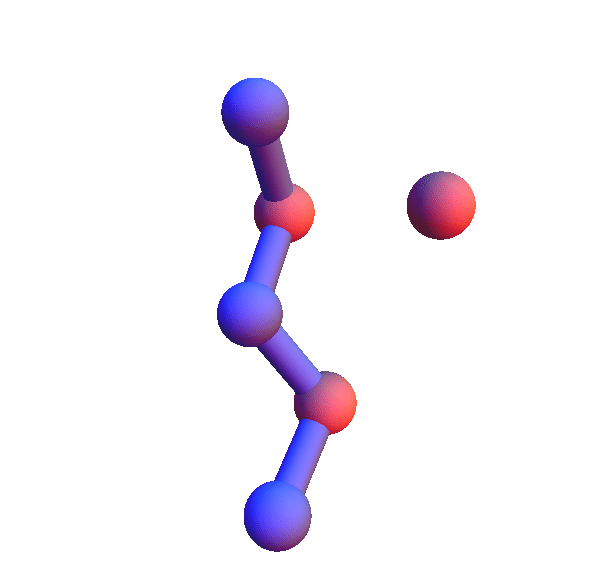
Not surprisingly many of the 6+1 structures form helices because that's a good way for two residues with backbone separation 3 to make a contact. But here is the structure of *L**K*:Y :
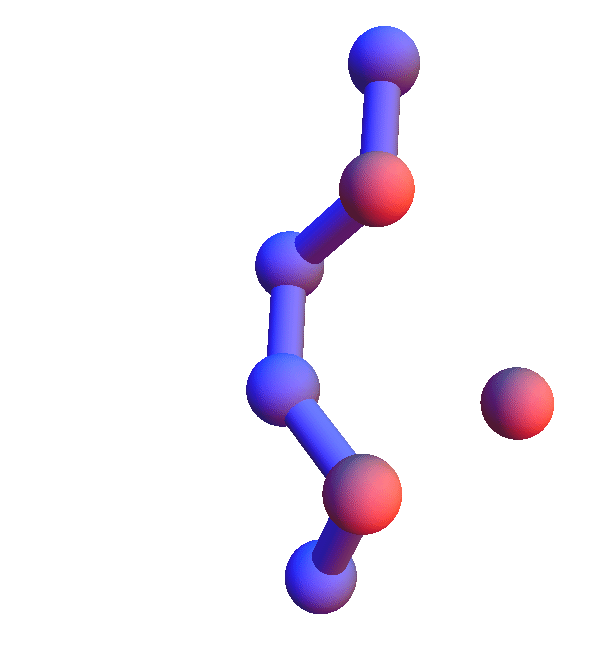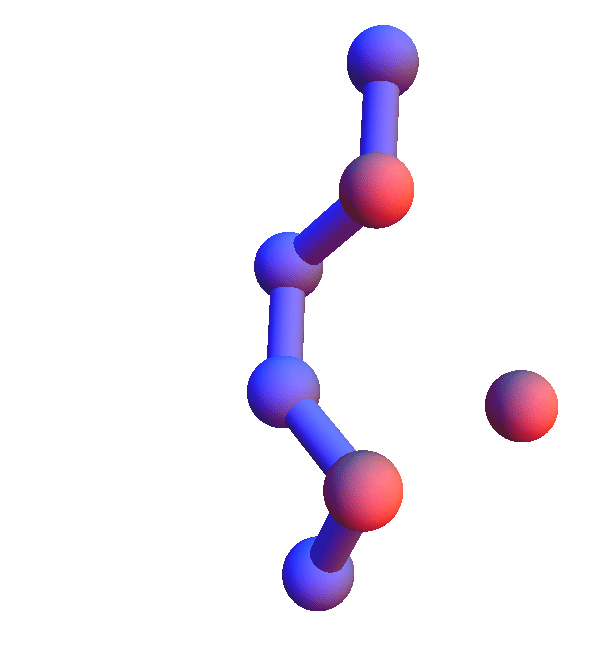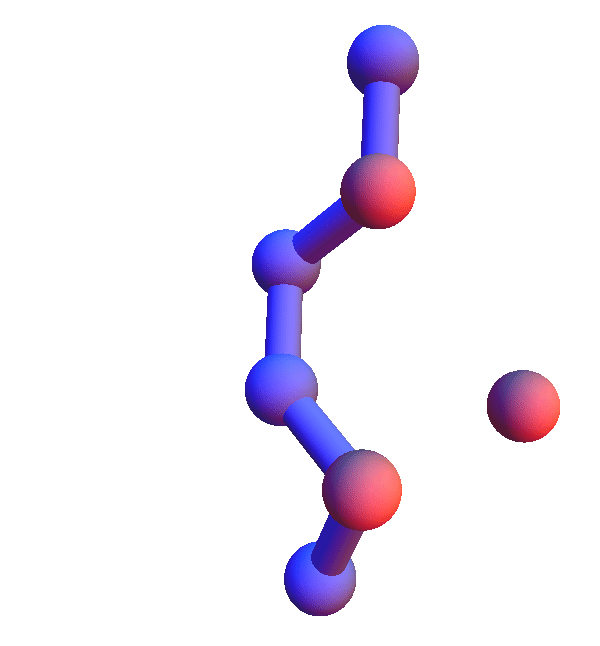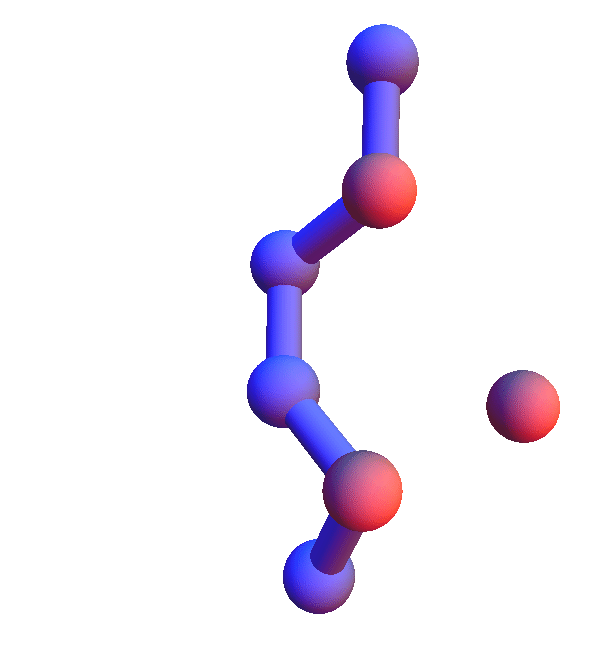
Thursday
No sightings of the fabled skunk cabbage on today's run, but I did get as far as 4 prisoners with Alex's puzzle. With that out of the way, let me turn to *L**L*:L, the hands-down most common local structure. That a trio composed of the most abundant residue in human proteins comes out on top is not surprising. Still, the popularity of the all-leucine trio is limited to the *X**Y*:Z codes; in the *X*Y*:Z codes it is in a multi-way tie with several codes where V and I are substituted for L, and in *XY*:Z codes L is strangely infrequent.
The *L**L*:L structures represent the limit of local structures at their most diverse. With two dihedral degrees of freedom per L, and no chemistry beyond the packing of methyl groups, there just are a lot of ways for three side chains to make mutual contacts!
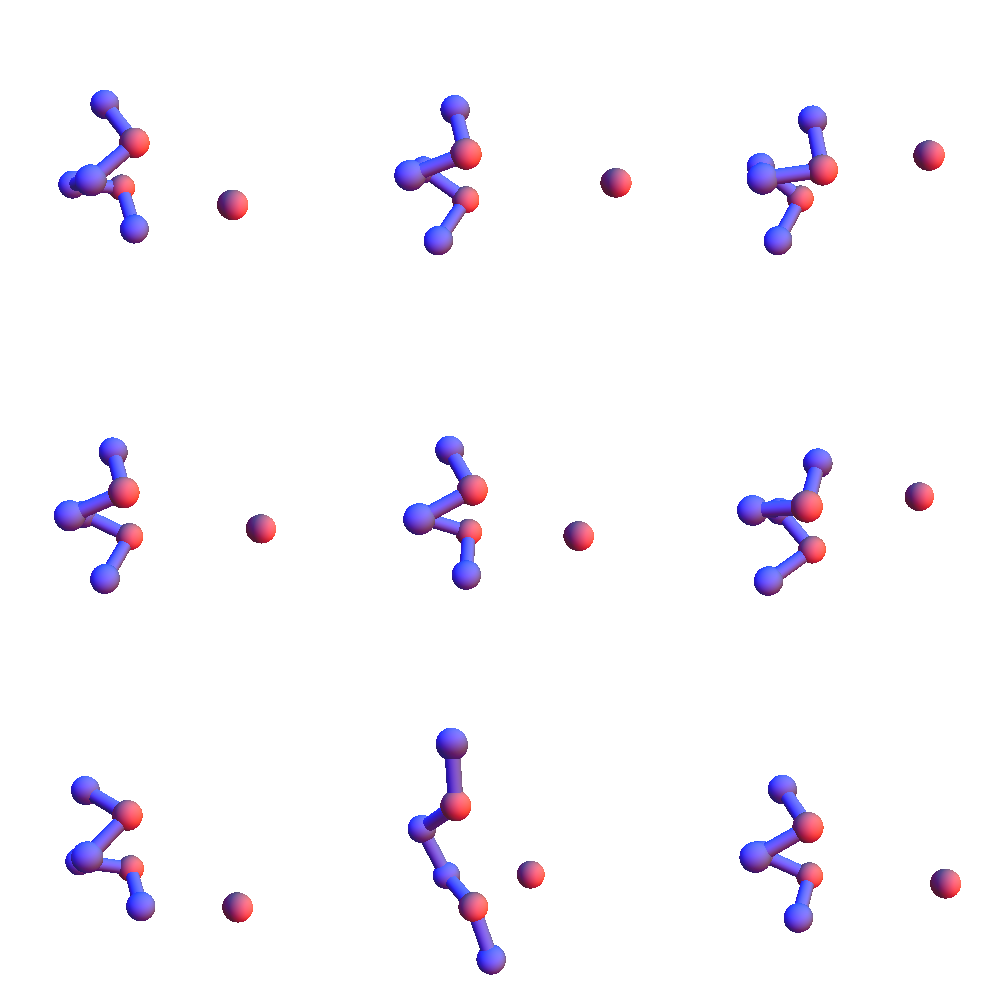
To get a handle on the diversity of *L**L*:L structures I generated a graph whose nodes are the 267 structures currently in my library, and where edges join nodes whenever the RMSD of the corresponding structures is within 0.5 Å. I then find a maximal independent set in this graph and use just the structures of those nodes to represent the variety of *L**L*:L structures. Here is a sampling of 9 of the 109 structures I obtained in this way:
I looked into the proposal of increasing the cutoff distance for atom contacts, so as to increase the number of triple-contact local structures. Recall (previous post) there was a curious peak at 5.5 Å in the distribution of the distance between nearest atoms in side-chain pairs. Turns out that peak is just an artifact, not a new type of contact. It often happens that the nearest atom pair of two side chains, when the residues are local on the backbone, are the atoms directly bonded to the α-carbons, the β-carbons. And since the α-carbon distance of adjacent residues is narrowly distributed, so will be the distance between the β-carbons. Conclusion: the largest contact distance is still that between two methyl groups, 4 Å. If anything, I should decrease my current 5 Å cutoff.
The low participation fraction (residues in trio-constraints) of a small minority of proteins, I've found, is explained either by very short chains (under 80 residues) or very non-compact folds, such as 4E40:
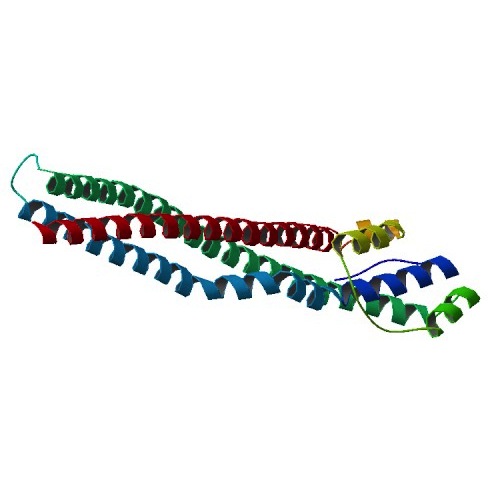
In either case, we have an unusually large surface-to-volume ratio. One option is to dismiss these oddball cases. The other option, that I'm favoring right now, is to add another type of local structure based on solvent contact.
Friday
While viewing various renderings of proteins in Mathematica I came to a startling conclusion: not all those red balls are residue oxygen atoms! In "BallAndStick" mode it's clear that the halo of red balls on the surface of the protein are the oxygen atoms of water molecules. At particular sites on the protein surface the hydrophilic interactions are sufficiently strong to localize water molecules, and these sites, to my delight, are well documented in the PDB! In my sample of PDB files, only a fraction of a percent do not include these water sites.
The availability of water sites presents an interesting opportunity. As this scatter plot shows,
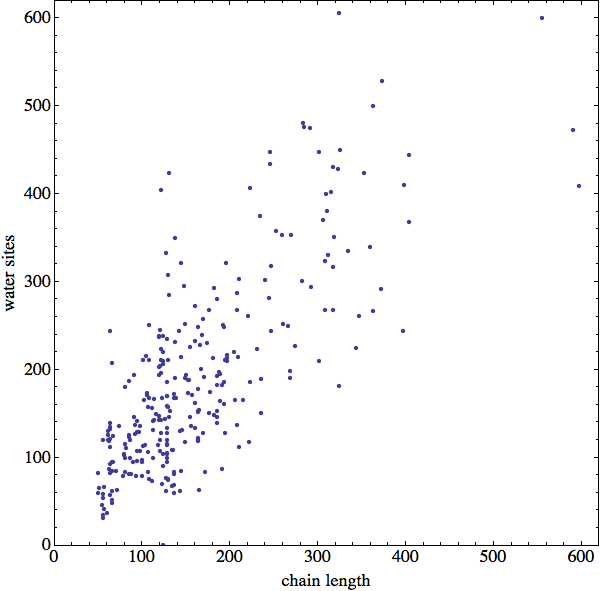
there are about as many water sites as residues in my sample of proteins. Thus, rather than try to model "solvent contact" constraints in some ad hoc way, why not build those constraints directly from the fine mesh of enveloping water sites that the PDB is kind enough to supply? I've put this at the top of my to-do list for next week.
