Submitted by ve10 on
I would like to start off by singing the praises of the number three. But first I need to bring up a point that was made abundantly clear over the past week: This project would go down in flames as a student thesis.
Cardinal rule 1 in supervising students is never having to tell them to start over, as in "from the very first thing you did". Students are primarily preoccupied with the execution of tasks and are working on the assumption that at least the premises that underlie those tasks are sound. And why shouldn't they be: the advisor dreamed them up!
Never being in the uncomfortable situation of asking a student to start from scratch is why I chose to do this project on my own. And that brings me to all the ways my original approach for representing local structure in proteins started unraveling last week.
Thursday
If I had to identify a core error in judgement it would be: faith that the sheer quantity of data in the PDB can make up for attention to physical detail. Even with "residue mutation neutrality" to partially collapse the dictionary of 20^5 residue subsequences, it was naive to think that a sufficiently powerful machine learning scheme would magically discover maps from local sequences to local structure. While such a thing cannot be ruled out, I needed evidence and I was not getting it.
The first goal I set was predicting local alpha-helix structure, without solvent contact, from a 5-residue sequence. This is by far the most common and most precisely defined local structure class. As a test of how well my local structure library was sampling this class, I applied residue point mutations (as derived from codon mutations) to see if these showed evidence of preserving the class. The result was that my library was woefully too small to even answer the question: essentially every mutation gave a new 5-residue sequence.
The mutation test tipped me off that local structure prediction was going to be very hard in my original scheme. I now had a choice: spend the next several months making incremental progress with ever more sophisticated machine learning tricks, or, revising the thing being learned. During my bus ride home I chose the latter.
Friday
The thing about protein secondary structure is that for the most part it is residue neutral. Residue side-chains are mere window dressing on a geometry determined by hydrogen bonding among just the peptide atoms. Because of this, predicting local structure from local sequence is bound to be elusive.
To promote residues to a level where their characteristics are more directly linked to structure, I defined a side-chain distance. The amino acid side-chain is the group of atoms, unique to each residue, that is attached to the α-carbon. My side-chain distance measure is the minimum distance over all atom pairs, one in each side-chain. Since the PDB structures for the most part do not include hydrogen atoms, only the C, N, O and S atoms are considered. The rationale for this distance is that it will be of order the methane diameter (4Å) no matter how exactly the two side-chains make contact (some residues having side-chains many times this size). Here is a histogram I made of all the side-chain distances under 8Å in a collection of 138 proteins:
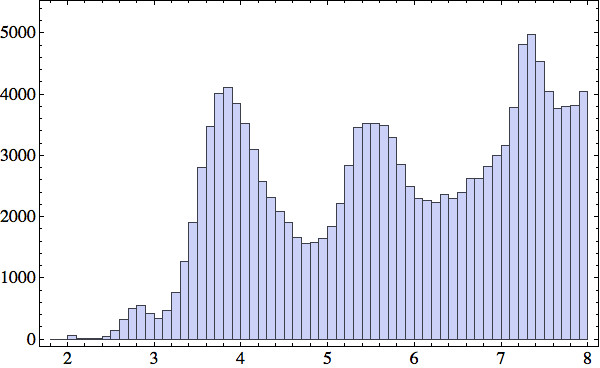
The peak at 4Å suggests that side-chains are well packed down to the atomic scale.
I've finally arrived at the divine triptych upon which my new local structure library is based: trios of mutually contacting residues. Here are three reasons why three is the right number:
1. Constraints based on the geometry of three points (the α-carbons associated with the contacting residues) are much stronger than two-point constraints.
2. Although 4-cliques (tetrahedra of α-carbons) would bring us even closer to a space filling description, these are too rare in proteins to be of much use.
3. The set of 20^3 possible "call numbers" looks like it may be small enough to sample in the PDB.
Looking ahead to how the local structures will be used in the constraint satisfaction algorithm, I am mostly interested in trios where at least two of the residues are local in the sequence (the residue order of the primary sequence is lost otherwise). Including only such trios, and setting the side-chain distance cutoff at 5Å (so as to include the aforementioned peak), I find that on average each residue is a participant in about two trios.
Saturday
My new structure library is starting to take shape. In addition to trimming my call numbers, I've also shortened my local α-carbon structures from five to four. Bear in mind that although the side-chains were consulted regarding structural contacts, in the end it is still the associated α-carbons that represent the fold.
There are eight types of local structure. In four of these all three contacting residues are local on the backbone: XYZ*, XY*Z, X*YZ, *XYZ. Here X, Y, Z are the residues making contact and * is a "wildcard" residue whose identity is not part of the structure's call number. Only about 6% of the library is completely local in this sense. In the other four structure types, the third residue (Z) is nonlocal on the backbone: X**Y, X*Y*, *X*Y, *XY*. I did not include XY** and **XY because I felt that the diversity of positions of wildcard α-carbons twice removed from a named residue would be too great.
