Submitted by ve10 on
It’s been a long summer, I realize, but that should not have diminished your interest in the resolution of the previous season’s cliffhanger ending. Can litemotifs fold proteins, or did the project run afoul of one of the three failure mechanisms sketched in the last installment?
But first a quick review of the status of the project as of May, 2014. The first version of the folding code, litemotif_1.0 (645 lines), is written and verified. I had verified the code by successfully folding a handful of short sequences, up to 100 residues, with “native” litemotif libraries. I say a library is native if all its litemotifs are derived from the target protein itself. Such libraries are not explicit blueprints for the fold: the algorithm still has to discover which contact atoms of a particular residue-type get matched up with each 4-residue subsequence. The much smaller size of native libraries speeds testing, such as the tricky divide-projection, where 5-atom groups are replaced by optimally aligned library motifs. It’s hard to imagine how one could get linear convergence (on a log scale) without every line of that projection-calculation code being correct.
There is a vast difference between a native library and the most complete library we can construct from the current-day PDB. Consider the protein 1I2T (human hyperplastic discs). Its native library comprises just 132 litemotifs, for an average of about two per residue. By contrast, the complete library holds 21,386 litemotifs that might apply to 1I2T. As the PDB grows, so will this number (but let’s not forget the premise behind the whole litemotif proposal, that is: litemotifs are indeed “motifs” and massively duplicated in the protein world). But the problem is not just that the folding algorithm runs about 160 times slower with the complete library, it also runs a higher risk of getting confused.
Before I undertook any serious experiments with complete libraries I upgraded the algorithm to take advantage of the spatial segregation of backbone and solvent. Real proteins rely on hydrophobic interactions to form compact shapes, and the algorithm should use this fact as a general constraint. The scheme I used in litemotif_2.1 (852 lines) is best explained with this cartoon:
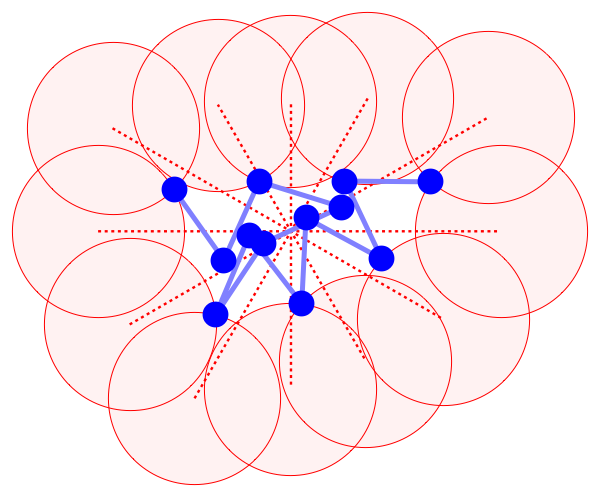
The segregation constraint is imposed just on the positions of the solvent atoms; the backbone atoms shown in blue are unconstrained. Solvent atoms are required to lie within spheres (shown in the cartoon as pink disks) of a given radius and centered on a set of rays. The rays originate from the centroid of the backbone and uniformly sample the space of directions. In 3D I use samples from a subdivision of the regular icosahedron. As the backbone conformation changes, so do the positions of the “bounding spheres”. I update the positions typically once every 100 iterations, moving each sphere inward along its ray just to the point where it first intersects a backbone atom.
Whereas litemotif_2.1 folds 1I2T even more efficiently, given its native library, when working with the complete library there is a serious problem. Below is a movie showing 20 frames spaced by 1,000 iterations (red balls are solvent):
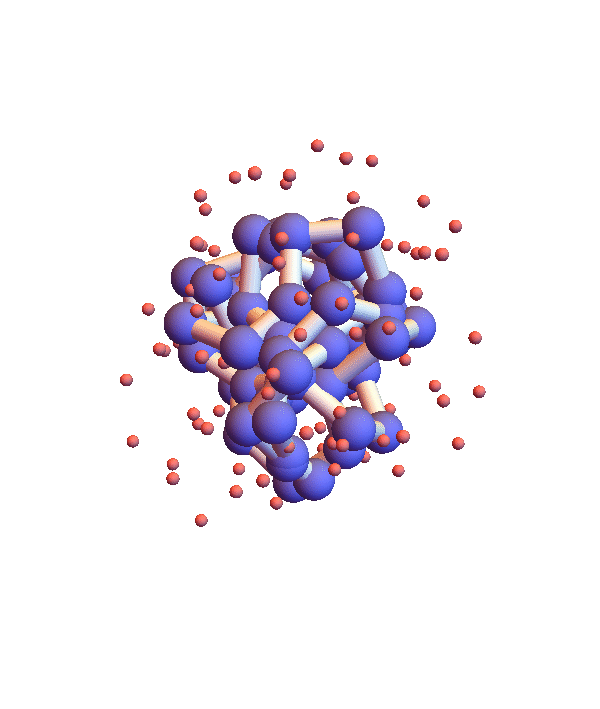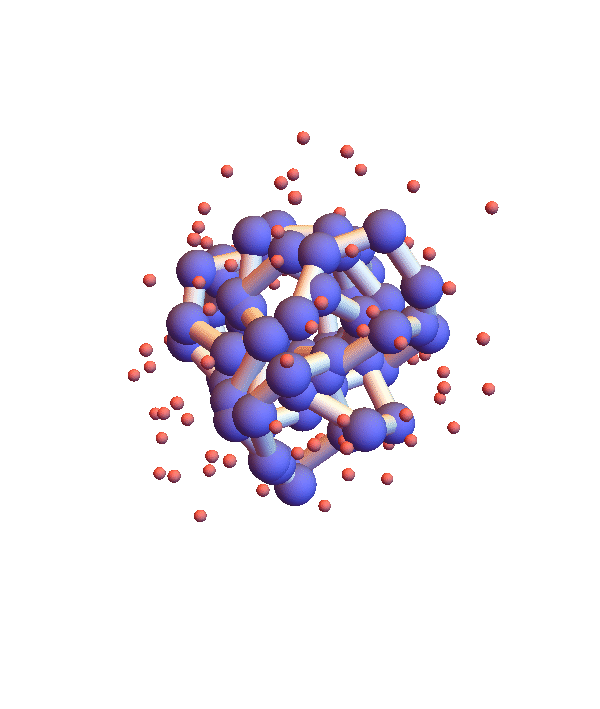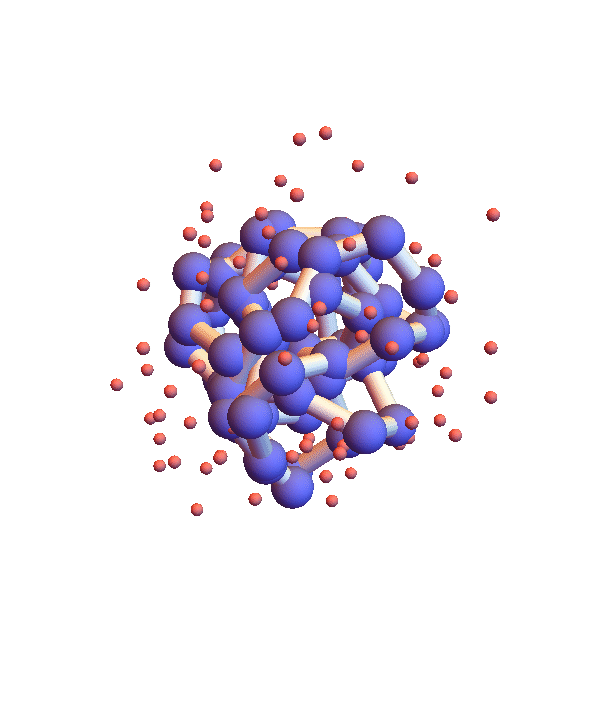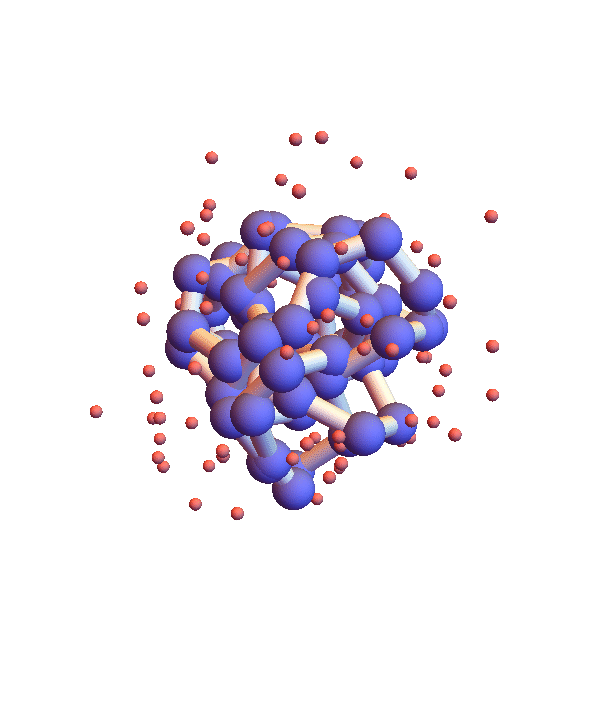
It seems the protein is clinging somewhat claustrophobically to the possibility of making many kinds of contacts, rather than exploring particular options. Of the speculated failure mechanisms of last May, the one that comes closest is, 3. combinatorial complexity. The algorithm knows it has not found the correct fold because the RMSD time series is still large, indicating that a large fraction of the constraints are unsatisfied. Below are two such time series, both using the complete library, but with different starting configurations, random and native-fold:
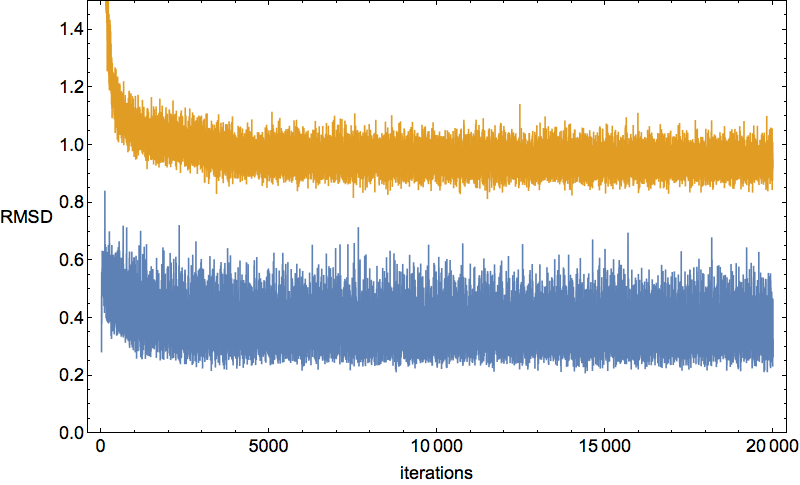
Apart from the fact that the random-start RMSD (yellow) is showing no signs of reaching the level of the native-fold-start (blue), you might be wondering why the latter is not exactly zero. There are two contributing factors. First, the native-fold start was not given the exact positions of the solvent atoms: it still has to discover their positions. The second contribution is perhaps even more significant, and the basis of how I plan to develop the algorithm in the next week.
The native library of 1I2T is missing litemotifs for eight of its 4-residue sequences. These occur at loops in the backbone, that is, where side chains are loosely packed and not recognized as contacts. But there are enough constraints on the rest of the backbone that this is not a problem. However, it turns out that there are proteins in the PDB that have these very same “loop subsequences” in the role of actual constraints. The folding algorithm does not know to ignore these constraints in the case of 1I2T, and as a result tries forcing it to do things it would rather not do!
Before describing my “selective ignorance” strategy for litemotif_3.0, I mention a simple enhancement I’ve already implemented as litemotif_2.2 (897 lines). The high density of backbone in the movie above is clearly unphysical and also can be addressed by a general constraint. In this case the trick is to express the constraint in such a way that the projection is still reasonably efficient and avoids the introduction of additional variables. The form of the constraint that works is “exclude from a sphere of a given radius, centered at the centroid of the motif, all backbone atoms other than those belonging to the motif”. Below is a movie showing the improvement one obtains when the exclusion radius is 4 Å:

Clearly not as dense and sluggish as in version 2.1, but still glacial for the task at hand. The only way to combat this stagnation, aside from modifying the algorithm, is to sample starting configurations. Below are four folds with 2.2, shown 100,000 iterations after different random starts:
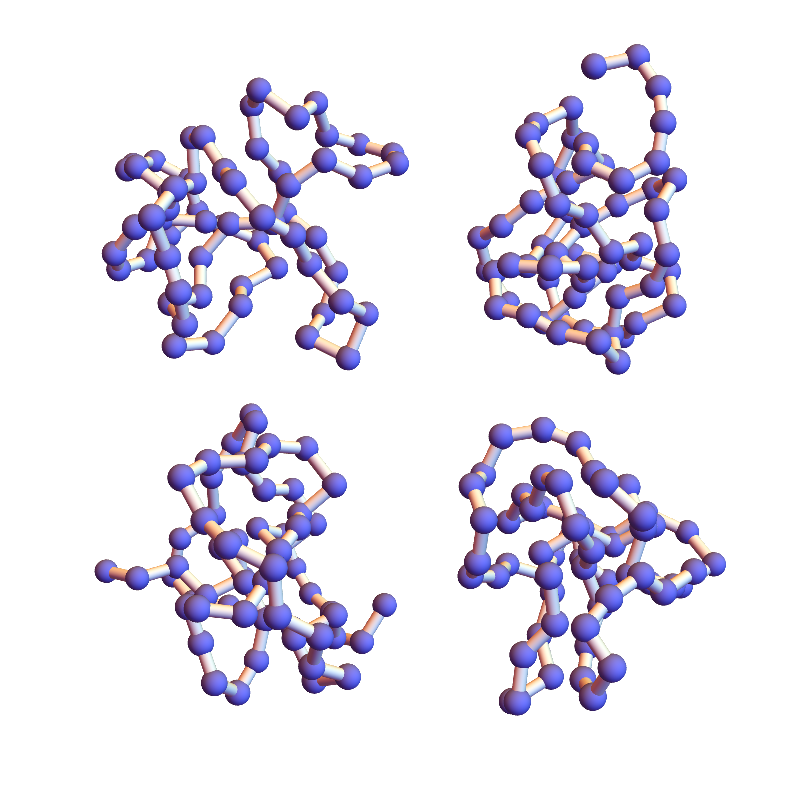
The final RMSD values of all of these is still significantly higher than what you get from the native-fold start.
These first experiments have shown that while litemotifs appear to contain enough constraints to single out the native fold, the algorithm is doing a terrible job exploring the space of folds. In version 3.0 I plan to test the hypothesis that the dynamics becomes more fluid when certain constraints along the backbone are ignored. Rather than concocting an ad hoc rule for deciding which constraints along the backbone can be ignored, say based on the numbers of litemotifs, my rule has more of a projection mind-set. The idea is simply that a given small number of the constraints are ignored. This is easy to implement as a projection, and should have the intended effect because the ignored constraints will be precisely those that are violated the most.
