Submitted by ve10 on
And no, it doesn’t dice. The scariest moment in a programming project, at least for me, is when I run the entire thing for the first time. Every component part has to be correct, not just in the logic of its design, but down to the finest details in execution.
I was lucky that litemotif.c produced output on the first run that looked like some approximation of what I was expecting. Often it takes me hours of debugging before I’m even at that stage.
A large share of the credit for the early launch goes to Pu Liu and Douglas Theobald, whose nicely documented Quaternion Characteristic Polynomial (QCP) package I used to perform the numerically most demanding computations. This is where optimal rotations are computed that map litemotif fragments from the PDB to the backbone of my protein. The calculation of these rotations, together with the easier translations, are in the innermost loop of the algorithm. It’s quite possible that during a single run of litemotif, the QCP function in the innermost loop is called more often than its combined usage by researchers worldwide doing more conventional things!
The “divide projection” PD, to litemotifs from the PDB, has to be one of the most exotic constraint-set projections ever attempted. But because this constraint is highly non-convex, an iterative algorithm built from PD, and the convex “concur projection” PC, is not expected to converge in a monotone manner. And that is putting it mildly.
The litemotif solver uses the difference-map iteration, also known by the name Douglas-Rachford. The update rule is very simple:
x’ = x + PC(2PD(x) - x) - PD(x).
The symbol x is the search vector: multiple copies of the positions of all the backbone and solvent atoms. When applied to NxN sudoku, the x in this iteration scheme is the four copies of the NxNxN grid of “cubicle occupancies” described in a previous post.
Much can be inferred about the nature of a problem from the behavior of what I will call the “RMSD time series”. The root-mean-square-deviation between x and x’ has two interpretations. First, its value represents the degree to which x is still changing and falling short of being a fixed point. Second, by the update formula we see that |x’ - x| is equal to the distance between two points we’ve found, one on each constraint set. Thus the RMSD value also represents the degree to which we’ve failed to satisfy both constraints.
It’s no big surprise that in sudoku, an NP-complete problem, the RMSD time series is highly erratic and reflects the chaotic dynamics of the search vector x. Here is a 1000-iteration segment generated by a hard 25x25 sudoku:
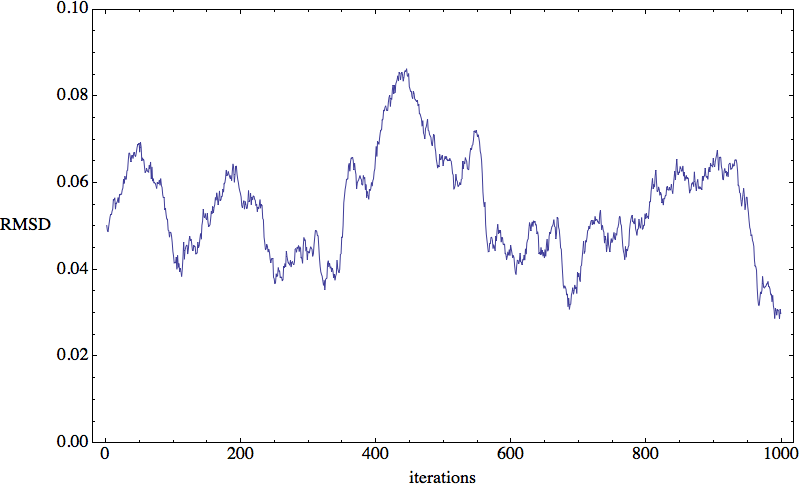
Nearly one million iterations are needed on average to solve this particular puzzle. Is folding proteins this chaotic?
Before answering that, let me first introduce the protein used to perform the critical test: 2P5K. This is a relatively compact 63-residue protein-fragment with sequence
NKGQRHIKIREIITSNEIETQDELVDMLKQDGYKVTQATVSRDIKELHLVKVPTNNGSYKYSL.
The litemotifs that apply to the 4-residue subsequences of 2P5K cover all the possible cases (solvent-only contacts, backbone-only contacts, solvent and backbone contacts, no contacts) and were therefore good for debugging. Here is the beginning of the contact list that litemotif.c reads as input:
NKGQ: O O
KGQR: D O O O O O
GQRH: Y O
QRHI: D O O
RHIK: L Y O
HIKI: D L O
IKIR: L O O
KIRE: D L O O
IREI: L L O O
REII: O O
EIIT: I L O
IITS: O O O O
ITSN:
TSNE: O O
SNEI: M
NEIE: I M N O O
etc.
The first line tells us there are two ways the first four residues (NKGQ) can make contact with solvent (O = oxygen), the next four residues can make contact with aspartic acid (D) or solvent (in five ways), etc. The subsequence ITSN makes no contacts (by my rules) and is probably loosely packed; no constraint will be applied to it. For each contact the input file has a set of 4+1 positions for the atoms participating in the constraint.
A relatively soft input parameter is the number of solvent atoms. This number is soft in the sense that not all of the solvent oxygens have to be used in constraints; those that are not, are bystanders and subject to no constraints at all. It is only the divide projection, PD, that chooses whether or not any particular solvent atom is used. If at some 4-residue sequence there is a choice between making contact with solvent, or a backbone atom, and the backbone-contact motif is nearer, then the solvent atom is ignored (for that part of the backbone). I could have used as few as 10 solvent atoms for 2P5K, but chose 45 thinking that would broaden the solution’s basin of attraction.
Because I am still only debugging, the litemotifs in my 2P5K input file were all derived from the known folded structure of this protein fragment. With this kind of input the algorithm has no excuse for not getting a perfect fold! However, once I start folding in earnest, the input data will be derived from all matching litemotifs in the PDB, and I will take special care to remove any that were derived from the specific structure being folded, or closely related ones, when they are known.
Now that you know what the test is being performed on, let’s see how folding proteins compares with solving sudoku. Here’s the RMSD time-series:
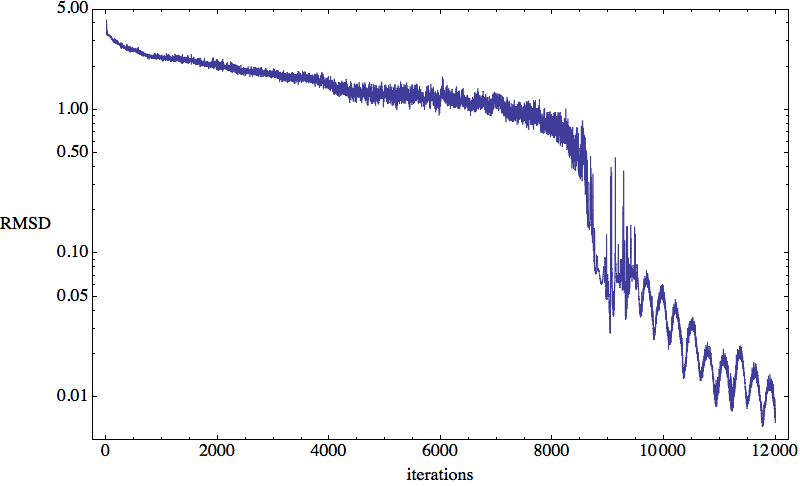
As you can imagine I ran a tiny victory lap as soon as I saw that steady march toward zero deviation (the vertical axis is logarithmic). This RMSD is normalized to be the root-mean-square deviation, on average, of the 58 5-atom litemotif structures that end up being used as constraints on the backbone; the units are Å. The time series shows that as soon as this number dips below about 1 Å, the search is essentially concluded. The peculiar oscillatory convergence that then ensues might be the result of tie-breaking among a pair of constraints that apply to the same 4-residue sequence, only one of which is used at any time.
How much actual searching is going on during those first 8000 iterations? Not that much, judging from an animation I made of the backbone. Here is a snapshot of the initial, random configuration that the program generates without any attempt at realism:

This quickly expands to the correct scale, followed by the different parts of the fold snapping into place:

As all the input litemotifs are consistent with the solution, the only searching that could be going on in this test is selecting the correct contact atoms. For example, if one of the Q(glutamine)-contact litemotifs is used, the algorithm is not told which of the four Q’s in the sequence is the correct one.
You’d think that this success has made me so woozy that I have to put off doing anything modestly technical for at least 24 hours. But this is hardly a consideration, given an even more significant event: today I’ve been married 30 years to the most remarkable aggregation of proteins ever to have appeared in the known universe. Of course you’re more than that sweetie — just trying to stay in character here.
